آموزش – تریتا پدیده صنعت
شرکت تریتا پدیده صنعت (TPS) متعهد است با تعریف، بیان و شفاف سازی مفاهیم و الزامات دارویی در اجرای سیستم کیفی به سازمان ها و شرکت ها کمک کند.

شرکت تریتا پدیده صنعت (TPS) متعهد است با تعریف، بیان و شفاف سازی مفاهیم و الزامات دارویی در اجرای سیستم کیفی به سازمان ها و شرکت ها کمک کند.

در طی دهه های اخیر، مشتریان اهمیت بیشتری به کیفیت محصولات میدهند. یکی از دلایل ممکن، شاید این باشد که مردم تمایل به اکتساب بهترین ها دارند. صنایع تولید دارو با رقابتی تنگاتنگ درون خودشان مواجه هستند. یکی از فاکتورهای مهم برای بقا در این صنعت، ایجاد و اجرای یک سیستم کیفی مناسب و کارآمد جهت کمک به تهیه داروهایی با اثر بخشی بیشتر و کیفیت بهتر است.
به دلیل ارسال و دسترسی مستقیم محصولات دارویی به بدن بیماران؛ ایمنی، خلوص، هویت و در نهایت کیفیت مناسب محصول، حائز اهمیت است. از این رو بحث مدیریت کیفیت در صنعت دارویی بسیار مهم و حیاتی میباشد. به همین دلیل تعداد فراوانی گایدلاین در سطح جهان نوشته و ترتیب داده شدهاند تا تمامی دستورالعملها و قوانین مورد نیاز در صعنت دارویی را پوشش دهند. سیستم مدیریت کیفیت به منظور حفظ کیفیت در محصولات دارویی تعبیه شده است.

ساخت و استفاده از محصولات دارویی که دربرگیرندهی اجزا سازندهاند، ضرورتا شامل درجاتی از ریسک هستند. ریسکی که بر کیفیت محصول وارد است تنها یک بخش کوچکی از تمامی ریسکهای موجود است. دانستن این نکته که کیفیت باید در طول عمر محصول حفظ شود بسیار حائز اهمیت است؛ چرا که رفتار و ویژگیهایش ( که نقش مهم و اساسی در کیفیت محصول دارند) باید ثابت باقی بمانند و شبیه به محصول استفاده شده در تحقیقات بالینی باشند.

آن دسته از داروهایی که با کیفیت پایین در بازار جهانی عرضه میگردند، به دلیل اضافه شدن ناخواسته یکسری از مواد سمی؛ نه تنها برای سلامت انسانها خطرناکاند، بلکه برای دولت و مصرفکننده نیز به نوعی هدررفت هزینه محسوب می شوند. بعنوان مثال درسال ۱۹۹۶در جزیرهی هائیتی، بیش از ۸۰ کودک براثر مصرف شربت سرفه و سرماخوردگی، که شامل گلیسرول آلوده به دیاتیلن گلایکول بود، جان خود را از دست دادند.

همانطور که در خصوص Warning Letters مطلع هستید، چنانچه مجموعهای به طور قابل ملاحظه الزامات سازمان غذا و دارو امریکا را رعایت نکنند، این سازمان در قالب نامهای تحت عنوان Warning Letters به مجموعه مرتبط هشدار میدهد. این نامه به صورت عمومی منتشر میشود تا سایر مجموعهها از نکات حاصل از آن درراستای جلوگیری از تکرار مجموعه خود استفاده کنند.

سازمان غذا و داروی آمریکا (FAD) در تاریخ ۲۸ تیر ماه ۱۳۹۸ (Jul.16.2019) در نامهای به شرکت Indoco Remedies Ltd. اعلام کرد که در یکی از نواقص این مجموعه، رسیدگی نکردن به عدم انطباقهای مشاهده شده در محصول است و از سوی دیگر اطلاعات در خصوص پروندههای تولید و کنترل کیفی از کفایت لازم برخوردار نیستند.
در این مقاله سعی برآن شده است تا با شرح یکی از نامه های اخطار که FDA آمریکا برای یک شرکت دارویی واقع در کانادا در آخرین ماه سال 2019 ارائه کرد، اهمیت به کارگیری سیستم CAPA در صنعت دارو را نشان دهیم.

با فرا رسیدن سال نو، کلیه افراد سعی در ایجاد تغییر در زندگی و روند پیشرفت خود دارند و سال نو رو بعنوان فرصتی مناسب برای شروع دوباره، برنامهیزی و از همه مهمتر مرور عملکرد خود در سال گذشته میبینند. چرا که تجربههای گذشته راهی برای پیشرفت در آینده هستند.
اهمیت این موضوع به قدری است که در صنعت داروسازی نیز مرور و بازبینی سالانه محصولات بعنوان یکی از الزامات رگولاتوریها مورد بررسی قرار میگیرد.
در طی دهه های اخیر، مشتریان اهمیت بیشتری به کیفیت محصولات میدهند. یکی از دلایل ممکن، شاید این باشد که مردم تمایل به اکتساب بهترین ها دارند. صنایع تولید دارو با رقابتی تنگاتنگ درون خودشان مواجه هستند. یکی از فاکتورهای مهم برای بقا در این صنعت، ایجاد و اجرای یک سیستم کیفی مناسب و کارآمد جهت کمک به تهیه داروهایی با اثر بخشی بیشتر و کیفیت بهتر است. بدین منظور مراجع قانونگذار در سراسر جهان، جهت اطمینان از تولید محصولات دارویی با کیفیت تضمین شده، راهنماهای سختگیرانه ای را وضع کردهاند. علیرغم ممارست های صنعتی، این رویکردها و راهنماها به ویژه برای تازه کارها شفاف نیستند. در این مقاله به مروری جامع از سیستم کیفیت دارویی توسط ICH Q10 پیشنهاد شده است در دیگر خطوط راهنما نیز شرح داده شده است میپردازیم. سیستم کیفیت دارویی دارای دامنه گسترده تر نسبت به GMP بوده و توسعه دارویی را نیز در برمیگیرد.
مفهوم سیستم مدیریت کیفیت دارویی (Pharmaceutical Quality System-PQS) در حال حاضر مبتنی بر راهنمای هماهنگ سازی بین المللی ICH Q10 و همچنین فصل اول راهنمای عملیات خوب ساخت PIC/S میباشد که در آن مدلی برای استفاده از رویکردهای مبتنی بر ریسک و دانش ارائه شده و میتواند در مراحل مختلف از چرخه عمر دارو با هدف ایجاد یک سیستم مدیریت کیفیت مؤثر برای صنعت داروسازی اجرا شود.
ICHQ10 شرح میدهد PQS یک سیستم مدیریتی با محوریت کیفیت، جهت هدایت و کنترل شرکتهای داروسازی است که تاکید می کند طراحی، سازماندهی و مستندسازی سیستم کیفیت دارویی باید شفاف و دارای ساختار مناسب باشد تا فهم کلی و کاربرد اصولی را تسهیل کند
در راهنمای PICS PE009-14 بخش اول اشاره شده است که دارنده ی یک پروانه بهره برداری باید اطمینان یابد که محصولات دارویی متناسب با اهداف مصرفشان تولید شوند، همچنین با پروانه ورود به بازار و مجوز کارآزمایی بالینی کاملا مطابقت دارند و بیمار را به دلیل نامناسب بودن از جهات ایمنی، کیفیتی و کارایی در معرض خطر قرار ندهند. جهت دستیابی به این هدف کیفی، باید یک سیستم کیفیت دارویی با طراحی جامع و اجرای صحیح و آمیخته با عملیات خوب ساخت، کنترل کیفیت و مدیریت ریسک کیفی (Quality Risk Management) موجود باشد.
تعریف WHO از سیستم کیفیت دارویی یک مفهوم گسترده است که تمامی جنبه هایی که به تنهایی یا در مجموع بر روی کیفیت یک محصول مؤثرند را پوشش میدهد. در یک سازمان تضمین کیفیت به عنوان یک ابزار مدیریتی عمل میکند.
راهنمای FDA اذعان میدارد که هر محصول دارویی جهت اطمینان از سطوح مورد نیاز ایمنی و اثربخشی، دارای هویت، قدرت، خلوص و سایر ویژگیهای کیفی طراحی شده است. هدف اصلی یک سیستم کیفی، تولید منسجم و یکپارچهی محصولاتی موثر و ایمن و حصول اطمینان از عملی بودن این فعالیتها میباشد. متخصصین کیفیت آگاه هستند که اهداف خوب به تنهایی ضامن تولید محصولات خوب نخواهند بود. تمامی بخش های یک سیستم کیفیت دارویی باید به میزان کافی از پرسنل شایسته، فضاهای مناسب و کافی، تجهیزات و امکانات بهره مند شوند. مفاهیم پایه مدیریت کیفیت، عملیات خوب ساخت و مدیریت بحران کیفیت به یکدیگر ارتباط دارند.
یک سیستم کیفیت دارویی باید اطمینان حاصل کند که:
1-آزادسازی محصول با طراحی، برنامه ریزی، نگهداری و پیشرفت مستمر سیستمی اتفاق میافتد که اجازه دریافت اصولی یک محصول با ویژگیهای کیفی مناسب را میدهد.
2- طراحی و توسعه ی محصولات دارویی برپایه ی مفاهیم و الزامات عملیات خوب ساخت (GMP) بنا شده است.

نمودار بالا، نشان دهنده ی ویژگی های اصلی سیستم کیفیت دارویی میباشد که توسط ICHQ10 ارائه شده است. PQS تمام چرخه ی عمر یک محصول را پوشش میدهد، که شامل پیشرفتهای دارویی، انتقال تکنولوژی، تولید محتوای تبلیغاتی و قطع تولید محصول میباشد. (همان گونه که در بخش بالایی نمودار نشان داده شده است.)
همچنین نمودار بیانگر این است که برای محصولات دارویی تحت بررسی ( تحقیقاتی) (Investigational Product) نیز، GMP مورد نیاز است.
خط افقی بعدی بیان کننده ی اهمیت مسئولیت های مدیریتی میباشد، که در بخش دوم در تمامی مراحل چرخه ی عمر محصول توضیح داده شد.
در خط افقی سوم به عناصر اصلی در مبحث PQS پرداخته شده است. این عناصر که نقش بنیادین و مهمی را بازی میکنند، باید بصورت کاربردی و مناسب در هر مرحله از چرخه ی عمر محصول اجرا شوند تا پیشرفت مستمر و توامان حاصل شود.
قسمت انتهایی نمودار نمایانگر توانمندسازها (Enablers) میباشد. مدیریت علوم و مدیریت بحران کیفیت، که در طول چرخه ی عمر یک محصول قابل اجرا هستند. توانمندسازها اهداف PQS را در جهت تحقق کیفیت محصول، ایجاد و حفظ حالت کنترل و تسهیل پیشرفت مستمر پشتیبانی میکنند.
بهبود مستمر یک سیستم کیفیت دارویی با کاربرد موارد زیر محقق میشود:
باید به این نکته توجه داشت که اجرای یک سیستم کیفی مؤثر در سازمان های تولیدی نیازمند سرمایه گذاری قابل توجه در زمان و منابع میباشد. اگرچه ما معتقدیم که مزایای اجرای یک سیستم کیفی در درازمدت بیشتر از هزینه های صرف شده خواهد بود.
در این بخش یک مدل از سیستم های کیفی قوی منطبق بر راهنمای FDA را شرح خواهیم داد، که چنانچه به طور صحیح اجرا شود، قادر خواهد بود کنترل هایی جهت تولید اصولی محصولی با کیفیت قابل پذیرش، فراهم آورد. این مدل براساس 4 فاکتور مهم توضیح داده شده است:
یک سیستم کیفی قوی با الحاق مکانیسم های دانش بنیان مؤثر به تصمیمات عملکردی روزانه ثبات فرآیند را ارتقا میبخشد.
در پایان هر دو عملیات خوب ساخت و عملیات خوب توزیع نیازمند یک سیستم کیفی قوی هستند. زمانی که یک سیستم کیفی به خوبی توسعه یابد و به شکل مؤثری مدیریت شود، به فرآیندهای یکدست و قابل پیشبینیای منجر میشوند که ایمنی، اثر بخشی و در دسترس بودن دارو و محصول را تضمین میکند (4).
منابع:
به دلیل ارسال و دسترسی مستقیم محصولات دارویی به بدن بیماران؛ ایمنی، خلوص، هویت و در نهایت کیفیت مناسب محصول، حائز اهمیت است. از این رو بحث مدیریت کیفیت در صنعت دارویی بسیار مهم و حیاتی میباشد. به همین دلیل تعداد فراوانی خطوط راهنما (گایدلاین) در سطح جهان نوشته و ترتیب داده شده اند تا تمامی دستورالعملها و قوانین مورد نیاز در صعنت دارویی پوشش دهند. سیستم مدیریت کیفیت به منظور حفظ کیفیت در محصولات دارویی تعبیه شده است.
سیستم مدیریت کیفیت درواقع مجموعه ای از فرآیند هایی است که تمرکز آن بر روی دستیابی به کیفیت ایده آل توسط القای خط مشی تنظیم مقررات و اهداف کیفی مطابق با نیازمندی های مشتریان میباشد. تصمیم استراتژیک یک سازمان، باید مطابقت با قوانین QMS باشد. طراحی و اجرای سیستم مدیریت کیفیت سازمان، متاثر از نیازهای مختلف، اهداف مشخص، تامین محصولات، فرآیند در حال اجرا، اندازه و ساختار سازمان میباشد.
مدیریت کیفیت در صنعت دارویی: اصول کلی و عناصر ضروری، مفاهیم بنیادی و عمومی تضمین کیفی را تعریف میکند. هم چنین که اجزای اصلی GMP و سیستم های زیر مجموعه اش (که تحقق آن مسئولیت مشترک بین مدیریت اصلی، بخش تولید و بخش کنترل کیفی است) را توضیح میدهد. این مفاهیم شامل: بهداشت، معتبرسازی، بازرسی ها، پرسنل، تجیهزات و فضاها، مواد و مستندسازی میباشد.
در صنایع دارویی گسترده، مدیریت کیفیت بعنوان یکی از جنبه های وظایف مدیریتی تعریف میگردد که خط مشی های کیفی را طرح و اجرا میکند. این سیاستها که شامل اهداف کلی و پیش برد سازمان با توجه به حفظ کیفیت است توسط مدیریت کل تعریف و تایید میگردد.
عناصر پایه ای مدیریت کیفیت شامل:
سیستم مدیریت کیفیت از چهار منظر قابل بررسی است:
یک سازمان دارویی با فهم درست و پیاده سازی سیستم مدیریت کیفیت مناسب قادر خواهد بود تا علاوه بر اخلاقیات، مسئولیت های قانونی شامل: مدیریت هویت، کیفیت، امنیت، خلوص و کارآمدی محصول نهایی دارو را، اجرایی کند. تمامی این مباحث نشان میدهد که اجرای سیستم مدیریت کیفیت مناسب، سبب دستیابی به یک صنعت معتبر میگردد.
منابع:
ساخت و استفاده از محصولات دارویی به همراه اجزاء تشکیل دهنده آن ها، دربرگیرنده ی درجاتی از ریسک هستند. ریسکی که بر کیفیت محصول وارد است تنها یک بخش کوچکی از تمامی ریسکهای موجود است. دانستن این نکته که کیفیت باید در طول عمر محصول حفظ شود بسیار حائز اهمیت است؛ چرا که ویژگیهای کیفی محصول (که نقش مهم و اساسی در کیفیت محصول دارند) باید در تمامی ساخت ها به صورت مداوم حفظ گردد و با ویژگی های کیفی محصول استفاده شده در تحقیقات بالینی منطبق باشند. رویکرد مؤثر مدیریت ریسک کیفی با فراهم نمودن ابزارهای فعال جهت شناسایی و کنترل مسائل بالقوه کیفی در طول توسعه و ساخت، قادر خواهد بود تا این اطمینان را ایجاد کند که کیفیت بالای محصول برای بیمار تحقق گردد. علاوه بر آن، استفاده از مدیریت ریسک کیفی میتواند قدرت تصمیمگیری را در زمان مواجه با مشکلات کیفیتی بهبود بخشد.
یک مدیریت ریسک کیفی موثر سبب میشود تا:
استفاده مناسب از مدیریت ریسک کیفی میتواند وظیفه صنعت را در انطباق با الزامات قانونی تسهیل بخشد اما هرگزاین تکلیف را حذف نخواهد کرد و خاطر نشان می گردد که جایگزین ارتباط مناسب بین رگولاتوری ها و صنعت نمیشود.
دو اصل مهم در مدیریت ریسک کیفی عبارتند از:
مدیریت ریسک کیفی یک فرآیند سیستماتیک است که به منظور ارزیابی، کنترل، ارتباط و مرور ریسکهای وارده به کیفیت محصول دارویی در طول چرخه عمر محصول، انجام میگیرد. یک مدل مدیریت ریسک کیفی در نمودار زیر نمایش داده شده است. میتوان از مدلهای دیگری هم استفاده نمود. تاکید بر هر جز از این چارچوب، ممکن است از موردی به مورد دیگر متفاوت باشد؛ اما یک فرآیند مستحکم، خواستار درگیری تمامی اجزا متناسب با یک ریسک مشخص در جزئیترین درجه است. یک نگاه کلی از مدیریت ریسک کیفی معمول، در تصویر زیر نشان داده شده است.

مسئولیتها:
فعالیتهای مدیریت ریسک کیفی به طور معمول، برعهدهی تیمی از افراد با رشتههای متنوع است. تیم شکل گرفته باید شامل متخصصانی از واحد های مرتبط مانند واحد کیفیت، مهندسی، امور نظارتی، تولید، توسعه تجارت، فروش و بازاریابی، آمار، حقوقی و بخش بالینی باشد. علاوه بر این افراد، کارشناسانی که دانش و احاطهی کامل به مباحث مدیریت ریسک کیفی دارند نیز باید در تیم حضور داشته باشند.
مراحل آغازی فرآیند مدیریت ریسک کیفی
مدیریت ریسک کیفی باید شامل طرحی از فرآیندهای سیستماتیکی باشد که هدف از طراحی آن ها هماهنگی، تسهیل و بهبود تصمیمهای برپایهی دانش (با نگاه به ریسک) است. مراحلی که برای آغاز و برنامهریزی مدیریت ریسک کیفی استفاده میشوند، ممکن است شامل موارد زیر باشد:
ارزیابی ریسک:
ارزیابی ریسک شامل تشخیص خطرات، آنالیز و ارزیابی ریسکهایی که در ارتباط با این خطرات هستند، میشود. ارزیابی ریسک کیفی با تعریف دقیق و مشخصی از مشکلات موجود و یا سؤالات ریسک آغاز میگردد. زمانی که ریسک در قالب سوال کاملا توضیح داده شد، ابزار مدیریت ریسک مناسب و نوع اطلاعات مورد نیاز به منظور ارجاع دادن به سوال ریسک، راحتتر قابل تشخیص خواهد بود. بعنوان راهنما، جهت تعریف شفاف از ریسکهای که در اهداف ارزیابی ریسک وجود دارند، معمولا سه سوال اصلی زیر مناسب است:
تشخیص ریسک یک استفادهی سیستماتیک از اطلاعات است تا از این طریق بتوان آسیبهای مرتبط با سوالات ریسک و یا شرح مشکلات را تشخیص داد.
آنالیز ریسک در واقع تخمینی است از ریسکهایی که در ارتباط با خطرات شناخته شدهاند.
مقایسهی بین ریسک شناخته و آنالیز شده و شاخصهای ریسک ارائه شده از جمله وظایف ارزیابی ریسک است. علاوه بر این، اعتبار و قدرت مدارک وشواهد را نیز برای هر سه سوال اصلی در نظر میگیرد.
خروجی ارزیابی ریسک میتواند در دوقالب تخمین کمیتی از ریسک و یا شرح کیفی از گسترهی ریسک باشد. زمانی که ریسک بصورت کمی بیان گردد، از احتمالات عددی استفاده میشود. در عوض، ریسک میتواند در فرم کیفی نیز با لغاتی همچون زیاد، متوسط و یا کم شرح داده شود. در این صورت باید تا حد المکان با جزئیات توضیحات ارائه گردد.
کنترل ریسک:
کنترل ریسک شامل تمامی تصمیم گیریها جهت کاهش و یا پذیرش ریسکها میشود. هدف اصلی کنترل ریسک، کاهش ریسک تا حد قابل قبول است.
کاهش ریسک:
کاهش ریسک بر روی فرآیندهایی تمرکز دارد که سبب کاهش و یا دوری از ریسک کیفی (زمانی که از سطح قابل قبول گذر می کند) میشود (به تصویر بالا مراجعه گردد). ممکن است کاهش ریسک شامل فعالیتهایی شود که شدت و احتمال آسیب رسانی را کاهش دهند.
پذیرش ریسک:
پذیرش ریسک تصمیمی است که ریسک را قبول میکند. پذیرش ریسک میتواند تصمیم formal به منظور پذیرش ریسکهای باقی مانده باشد و یا تصمیم passive باشد که در اینصورت ریسکهای باقیمانده تعیین شده نیستند.
ارتباط ریسک:
ارتباط ریسک درواقع اشتراک گذاری اطلاعات ( دربارهی ریسک و مدیریت ریسک)، بین تصمیم گیرندهها و دیگران است.
مرور ریسک:
مدیریت ریسک در واقع یک بخش همیشه در حال اجرا از فرآیند مدیریت کیفی است. یک مکانیسم به منظور مرور و پایش اتفاقات باید اجرایی گردد.
خروجی و یا نتیجهی مدیریت ریسک کیفی باید بازبینی شود تا دانش و تجارب جدید در نظر گرفته شوند. هنگامی که مدیریت ریسک کیفی آغاز شد، این فرآیند باید تا حدی ادامه پیدا کند تا برای تمامی اتفاقاتی استفاده شود که ممکن است بر تصمیمات مدیریت ریسک کیفی اصلی تاثیر بگذارند. اتفاقات میتوانند برنامهریزی شده باشند (مانند نتایج مرور محصول، بازرسیها، بازبینی ها، کنترل تغییرات) و یا آن دسته که برنامه ریزی نشدهاند (مانند فراخوان و علل ریشهای که از نتایج تحقیقات شکست خورده بدست آمدهاند). تکرار هرگونه بازبینی باید برپایهی درجهی ریسک باشد. بازبینی ریسک ممکن است شامل تجدید نظر در تصمیمات پذیرش ریسک باشد.
علاوه بر این، صنایع دارویی و رگولاتوریها میتوانند ریسکها را با استفاده از ابزارهای تشخیصی مدیریت ریسک و یا روشهای داخلی ( مانند روشهای انجام استاندارد) ارزیابی و مدیریت کنند. در زیر به تعدادی از این ابزار اشاره شده است:
مدیریت ریسک کیفی باید تمام هدفش را بر روی افزایش سطح محافظت از بیمار متمرکز کند. این هدف زمانی نائل میگردد که ریسکی که بیمار در زمان مصرف مستقیم دارو با آن مواجه میشود، کاهش یابد.
فعالیتهای بسیاری به منظور بهبود کیفیت محصول دارویی انجام گرفته است و امروزه با چارچوب رگولاتوری در یک راستا حرکت میکنند. اما هنوز هم مسائل بسیاری در این زمینه وجود دارند که نیازمند توجه هستند. QRM کمک میکند تا ریسک وارده بر بیمار و شرکت تولید کننده را مدیریت کند. مشکلات مختلفی در حوزهی ساخت در زمان آزادسازی بچها یا پیش از آن هنوز هم دیده میشوند؛ که سبب میشود تا تحقیقات هزینه بر و پیچیده و دیگر عیوب جدی کیفی به وقوع بپیوندند. تمامی اینها درنهایت، باعث فراخوان محصول و توقف تولید بچها میگردد. ثمرهی اصلی اجرای QRM در ساخت محصولات دارویی، دستیابی به داروهای ایمنتر برای بیماران است. علاوه بر این سبب ایجاد یک رویکرد مقرون به صرفه و کارآمد برای احراز کیفیت، معتبرسازی، کنترل تغییرات و دیگر حوزههای کنترل کیفیت میگردد.
منابع:
آن دسته از داروهایی که با کیفیت پایین در بازار جهانی عرضه میگردند، به دلیل اضافه شدن ناخواسته یکسری از مواد سمی؛ نه تنها برای سلامت انسانها خطرناکاند، بلکه برای دولت و مصرفکننده نیز به نوعی هدررفت هزینه محسوب می شوند. بعنوان مثال درسال ۱۹۹۶در جزیرهی هائیتی، بیش از ۸۰ کودک براثر مصرف شربت سرفه و سرماخوردگی، که شامل گلیسرول آلوده به دیاتیلن گلایکول بود، جان خود را از دست دادند. اگر در زمان ساخت شربت، سازنده تمام اصول عملیات خوب تولید را رعایت مینمود، میتوانست از تمامی این مرگ و میرها جلوگیری به عمل آورد. مضاف بر این، دارویی که فاقد مواد تشکیل دهنده ادعا شده در لیبل دارویی باشد یا مقدار کمتری از مقادیر اعلام شده را داشته باشد، مسلما تاثیر درمانی مطلوب را نخواهد داشت. به عنوان مثال آنتی بیوتیکی که میزان مادهی فعالش کمتر از حد درمانی آن باشد، قابلیت درمان عفونت را نخواهد داشت. شرایط بدتر زمانی اتفاق میفتد که باکتریها درمواجهه با سطح پایین آنتیبیوتیک، حتی درصورت درست بودن دوز درمانی تجویز شده، از بین نمی روند و درمقابل دارو مقاومت پیدا می کنند. تمامی این اتفاقات تاثیر مستقیم بر سلامت انسان میگذارد و زندگی بسیاری را به خطر میاندازد.
سازمان بهداشت جهانی WHO، عملیات خوب تولید را بعنوان آن بخش از تضمین کیفیت تعریف می کند که یکنواختی و ثبات تولید محصول و کنترل مداوم آن را مطابق با استانداردهای کیفی مربوطه ضمانت میکند. این استانداردهای کیفی باید متناسب با موارد استفاده محصول و مطابق با مجوز فروش باشد. GMP تمامی بخشها و ابعاد فرآیند ساخت را پوشش میدهد؛ من جمله: فرآیند ساخت تعریف شده، مراحل ساخت حساس و بحرانی که معتبر شدهاند؛ تجهیزات، انبارداری و انتقال مناسب؛ پرسنل تولید و کنترل کیفی آموزش دیده و با صلاحیت؛ امکانات آزمایشگاهی مناسب؛ روشها و دستورالعملهای نوشته که تایید شده؛ ثبتنامه هایی که نشاندهندهی تمامی مراحلی هستند که دریک پروسهی تعریف شده انجام گرفته است؛ قابلیت ردیابی کامل محصول از طریق مستندات ساخت یک بچ در طول فرآیند ساخت و مستندات توزیع؛ و سیستمهایی که برای فراخوان مرجوع و بررسی شکایات استفاده میشود. اصول راهنمای GMP بیان میدارد که کیفیت در طول ساخت یک محصول ایجاد میشود نه فقط در آنالیز محصول نهایی. بنابراین، تضمین کیفیت این است که محصول نه تنها، محدوده های مرحلهی نهایی را دارا باشد بلکه اطمینان خاطر بدهد که محصول نهایی هربار تحت شرایط یکسان و فرآیندهای یکسان ساخته میشود. راههای زیادی برای کنترل این مورد وجود دارد؛ کنترل کیفیت امکانات و سیستم های موجود؛ کنترل کیفیت مواد اولیه، کنترل کیفیت تولید در تمام مرحله، کنترل کیفیت تست های آنالیز محصول، شناسایی و کنترل هویت محصول بواسطهی لیبل زدن و جدا سازی از طریق فضاهای مختلف، کنترل کیفیت مواد و محصول نهایی بواسطهی انبارداری مناسب و غیره. تمامی این کنترلها باید طبق روشهای تعیین شده، رسمی و تصویب شده، پروتکل های نوشته شده، روش های کار استاندارد و Master Formula، توصیف کننده تمام مراحل انجام شده در کل فرآیند ساخت و کنترل محصول باشند.
GMP از خطاهایی پیشگیری میکند که قادر به حذف در طول فرآیند کنترل کیفی محصول نهایی نیستند. حصول اطمینان از یکسان بودن کیفیت تمامی واحدهای یک دارو با آن چه در آزمیشگاه تست شده، بدون GMP غیر ممکن به نظر میرسد. در اوایل دههی ۱۹۷۰، شرکت سازندهای در ایالات متحدهی بریتانیا محلول تزریقیای را تولید نمود که بدلیل آلودگی بالای باکتریایی، سبب مرگ پنج بیمار شد. قبل از توزیع این محلولهای تزریقی، طبق تستی که سازنده بر روی تعداد زیادی از بطریها انجام داده، تمامی آنها استریل بودهاند. اما در طی تحقیقاتی که در جهت یافتن علت مرگ بیماران صورت گرفت، مشخص شد که خطای تکنیکی در بخش استریلیزاسیون بوده است. به این دلیل که بطریهای موجود در بخش پایینی دستگاه به درستی استریل نشده بودند و بطریهای تست شده از سمت سازنده همه از بخش بالایی بودهاند که باعث نتیجه گیری اشتباه از استریل بودن تمام بچ شده است.
امروزه تطابق با GMP برای دریافت پروانهی ورود به بازار جز شرایط ضروری بحساب میآید. به عبارتی دیگر تولید کنندههای خارجی و داخلی صنعت دارو، بدون داشتن GMP قادر به فروش و یا بازاریابی محصولاتشان در داخل یا خارج کشور نخواهند بود. این درحالی است که تطابقات GMP هنوز به شکل گسترده با جهان در حال توسعه سازگار نشده و بیشترین فشار بر روی دولتهایی است که در کشورهای کمتر توسعه یافته هستند؛ چرا که حتی دریافت پروانهی ورود به بازار در بخش داخلی نیز کار دشواری بنظر میرسد. از سمت دیگر، کشورهای خارجی نیز استراتژیهای مختلفی به منظور حصول اطمینان از سازگاری کشورهای درحال توسعه با قوانین GMP ایجاد کرده است. اجرای الزامات GMP، نیازمند یک سرمایه گذاری عظیم در زمینهی ارتقای امکانات ساخت است و به طبع چنین سرمایه گذاری برای تولید کنندهی داخلی، پیامدهایی گزافی خواهد داشت. سوالی که در اینجا مطرح میشود، تاثیر این تغییرات بر روی بازار داخلی و دسترسی به دارو و تامین آن در کشوهای درحال توسعه می باشد.
اغلب الزامات GMP به منظور پاسخگویی در شرایط بحرانی و یا جلوگیری ازوقوع موقعیتهای خطرناک در آینده ایجاد شدهاند. در جهت ایجاد و حفظ چنین الزاماتی، تمامی مدیران و سوپروایزرین باید بستری را فراهم سازند تا از انجام یک خود بازرسی GMP مداوم و معنیدار، کارکنانی آموزش دیده و متخصص و مشارکتی کامل در برنامههای در حال اجرا اطمینان حاصل کند. مدیریت سازمان باید بصورت رسمی و عمومی از طریق عملکرد خود اعلام نماید که تنها راه حضور در بازار دارویی مطابقت با استانداردهای GMP می باشد.
به منظور پیش روی و پایبندی مردم با اصول GMP، نیاز است تا علت پدیدآمدن الزامات، اهمیت الزامات برای هرکسی به عنوان مصرف کننده را بدانند تا از آنها پیروی کنند.
مراجع:
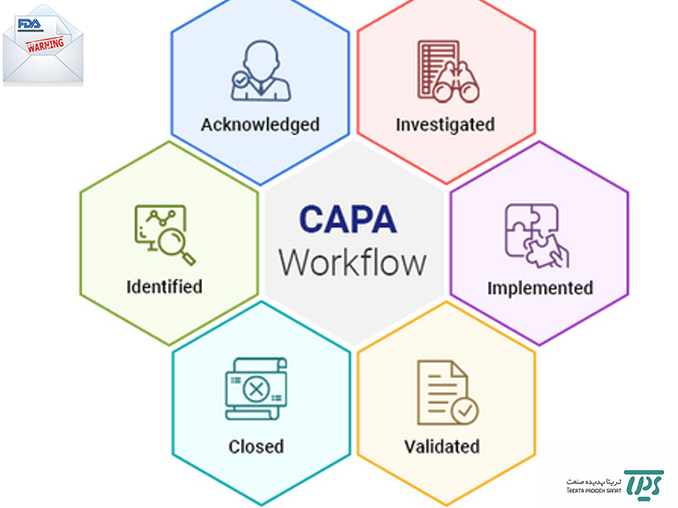
همانطور که در خصوص Warning Letters مطلع هستید، چنانچه مجموعهای به طور قابل ملاحظه الزامات سازمان غذا و دارو امریکا را رعایت نکنند، این سازمان در قالب نامهای تحت عنوان Warning Letters به مجموعه مرتبط هشدار میدهد. این نامه به صورت عمومی منتشر میشود تا سایر مجموعهها از نکات حاصل از آن درراستای جلوگیری از تکرار مجموعه خود استفاده کنند. در همین راستا مجموعهی تریتا پدیده صنعت خلاصهای از نامههایی که حاوی آیتمهای حائز اهمیت و آموزنده هستند را به همراه نکات استخراج شده تحت عنوان Warning Letters & Lessons Learned در اختیار همراهان این مجموعه قرار میدهد.
سازمان غذا و داروی آمریکا (FAD) در تاریخ ۲۸ تیر ماه ۱۳۹۸ (Jul.16.2019) در نامهای به شرکت Indoco Remedies Ltd. اعلام کرد که در یکی از نواقص این مجموعه، رسیدگی نکردن به عدم انطباقهای مشاهده شده در محصول است و از سوی دیگر اطلاعات در خصوص پروندههای تولید و کنترل کیفی از کفایت لازم برخوردار نیستند. نکات قابل استخراج از متن نامه بصورت اختصار در زیر آورده شده است: 1.ضعف از یکپارچگی اطلاعات ( Data Integrity) به عنوان یکی از نواقص ریشهای مطرح شده، که خود منجر به نواقص دیگر میشود 2.توصیهی سازمان غذا و دارو، بهره گرفتن از مشاورانی است که در مبحث یکپارچگی اطلاعات صاحب صلاحیت باشند (Highly qualified consultant in data integrity) 3.همچنین تاکید شده که استفاده از مشاور، از مسئولیت مدیریت اجرای مجموعه (firm’s execution manager) نمیکاهد و مسئولیت ایشان در حل عدم انطباقها از منظر قانونی به عهدهی خودشان است 4.در خصوص محصولاتی که هنوز وارد بازار نشدهاند هیچگونه توجیهی در عدم رسیدگی یا تعویق در رسیدگی به عدم انطباقها پذیرفته نیست و مطابق با زمانبندیهای تعیین شده، این رسیدگی میبایست در دستور کار قرار گیرد
CAPA نقش مهمی در سیستم مدیریت ریسک ایفا میکند؛ چرا که منجر به پیشرفت محصول و فرآیند و درک محصول و فرآیند پیشرفته میشود. با اجرای صحیح فرآیند CAPA، میتوان با جمع آوری اطلاعات ، تجزیه و تحلیل ، شناسایی و بررسی مشکلات مربوط به کیفیت محصول و انجام اقدامات پیشگیرانه/اصلاحی مؤثر، از وقوع موارد مشابه یا بروز مجدد انحرافات/عدم انطباقات جلوگیری نمود.
در این مقاله سعی برآن شده است تا با شرح یکی از نامه های اخطار که FDA آمریکا برای یک شرکت دارویی واقع در کانادا در آخرین ماه سال 2019 ارائه کرد، اهمیت به کارگیری سیستم CAPA در صنعت دارو را نشان دهیم.اخطارنامه شماره ۱۵-۲۰-۳۲۰، ۰۲/۱۰/۱۳۹۸:با توجه به بازرسی FDA در سپتامبر 2019 از شرکت مذبور و ارائه گزارش مبنی بر مشاهدهی عدم انطباق با الزامات رگولاتوری در خصوص عملکرد، حفظ سوابق کتبی کافی از کالیبراسیون تجهیزات، بازرسی تجهیزات اتوماتیک، الکترونیک و مکانیک که در ساخت، فرآیند، بستهبندی و نگهداری محصول دارویی(21 CFR 211.68 (a)) کاربرد دارند، سازمان غذا و دارو امریکا از شرکت مذکور درخواست ارائه CAPA جهت برطرف کردن مشکلات موجود کرد. این شرکت CAPA را تهیه و ارسال نمود؛ اما پاسخ و اقدامات در نظر گرفته، مورد قبول سازمان غذا و دارو امریکا نبود. در اخطارنامه قید شد که جزئیات کافی و شواهد مربوط به افدامات اصلاحی مبنی بر اجرای صحیح فرآیندها منبطق بر اصول CGMP کافی و مناسب نبوده و مورد پذیرش این سازمان نیست. یک نمونه از نقص های موجود در پاسخ این شرکت به شرح زیر است:در پاسخ ارائه شده از سوی این شرکت، بیان شد که آنها سوابق مربوط به بچهای محصول را به طور تصادفی برای مقایسه با دادههای HMI، انتخاب میکنند؛ سپس مغایرتهای بین سوابق بچ محصول و دادههای HMI جمع آوری شده را مقایسه مینمایند. FDA اظهار داشت که شرکت مذکور تعهد کافی به ارزیابی کامل مغایرتها و عدم انطباق بین دادههای HMI و سوابق بچ را ندارد و به نظر میرسد که این شرکت دلایل ریشهای مربوط به این عدم انطباقات را مشخص نکرده است. هم چنین تحقیق و بررسیهای لازم به منظور تشخیص انحرافات/عدم انطباقات بالاقوه در سایر نواحی تولید انجام نشده و ارزیابی و اثربخشیهای لازم بر محصول نهایی را ارائه نکرده است. علاوه بر این، شرکت مذکور، به شکست مدیریت عملیات و عدم نظارت کافی واحد کیفی بر مستندات و بررسی یکپارچگی ها آنها نپرداخته است. در این نامهی اخطار، پس از ارائه دو عدم انطباق دیگر ، مبنی بر عدم پاسخگوی مناسب و اقدام اصلاحی موردنیاز، مسئول انطباق بر اصول GMP در سازمان FDA، به شدت توصیه نمود تا به منظور اجرای صحیح الزامات مندرج در CFR 211.34، از مشاور واجد شرایط و مشرف بر این قوانین کمک و مشورت گیرد تا سبب انطباق هر چه بیشتر شرکت با الزمات CGMP جهت ورود به بازار داروهای امریکا گردد.
چرا که تجربههای گذشته راهی برای پیشرفت در آینده هستند.
اهمیت این موضوع به قدری است که در صنعت داروسازی نیز مرور و بازبینی سالانه محصولات بعنوان یکی از الزامات رگولاتوریها مورد بررسی قرار میگیرد.
به همین روی در این مقاله سعی داریم تا به یکی از اصلیترین مسائل مطرح شده در اصول عملیات خوب ساخت با تکیه بر راهنمای PIC/S بپردازیم.
PQR چیست؟
APR یا PQR دو واژهای هستند که هر دو یک مفهوم کلی را در بردارند و عبارتاند از مرور و بازبینی کلیه مدارک و مستندات مربوط به یک محصول با هدفهای مشخص.
در ابتدا بدانیم که PQR مخفف واژهی Product Quality Review میباشد و عموماً در رگولاتوریهای بخش اروپا کاربرد دارد؛ در حالی که APR مخفف واژهی Annual product review میباشد که در رگولاتوریهای امریکا به این نام خوانده میشود.
همانطور که از مخفف این واژه مشخص است معمولا بصورت سالانه انجام میگیرد اما لزومی ندارد که تعیین سال با شروع سال نو باشد.
هدف از مرور سالانهی اسناد مربوط به یک محصول، اطمینان از تداوم فرآیند، تشخیص عیوب کیفیتی، نقص در سیستم و فرآیند، بررسی تغییرات مورد نیاز در مشخصات، فرآیند تولید و ساخت و روشهای کنترل و ارزیابی، لزوم معتبرسازی مجدد میباشد.
بازبینی کیفیت محصول عموماً در جهت تعیین اقدامات اصلاحی به منظور بهبود بیشتر طراحی میشود و اهداف زیر را به دنبال دارد:
PQR بایستی حداقل شامل موارد زیر باشد:
مرور بر مواد اولیه (مواد اولیه شامل مواد جانبی و مواد موثره)
مرور بر کنترلهای حین تولید و مرور بر نتایج محصولات نهایی
مرور بر بچهای تولیدی، میزان بچهای بازگشت خورده و دلیل مربوط به آنها
مرور بر بچهای که مشخصات لازمه را پاس نکرده بودند و تحقیق وبررسیهای مربوط به آنها
مرور بر اثربخشی اقدامات اصلاحی
مرور بر شکایات مربوط به کیفیت محصول و مرجوعیها
مرور بر کلیه انحرافات/ عدم انطباقات
مرور بر تغییرات وارد بر فرآیند و یا روشهای آنالیزی
مرور بر نتایج مطالعات پایداری
گزارش PQR
گزارش PQR حداقل شامل موارد زیر میباشد:
ارزیابی نتیجهی PQR
سازنده میبایست نتیجهی PQR را ارزیابی نموده و اقدامات اصلاحی جدید جهت رفع مغایرتها و یا هرگونه معتبرسازی مجدد را تهیه و ابلاغ نماید.